重なったシークエンスの波形を分離する
CRISPR-Cas9で遺伝子の破壊は容易にできるようになりました。シロイヌナズナの遺伝子を破壊する場合、形質転換をして最初に得られた種子(T1)世代では相同染色体のそれぞれに起きた変異が異なる状態(biallelic)になっていることが多いです。この変異をシークエンスによって調べようとすると、長さの異なるものがあるので、きれいに読むことができません。
ちゃんと調べるにはホモ接合体をとるか、PCR産物をクローニングしてシークエンスをする必要があるのですが、いずれにせよ時間がかかります。この重なった波形をうまく分離できればいいのですが、人の手でやるとかなり大変です。そこで、Mitsucalに補助プログラムを実装してみました。
サンプルデータ
以下の説明手順に使用したファイルと塩基配列です。
サンプルファイル(ab1)
シークエンスした範囲の塩基配列
手順
- Mitsucalの仮想実験室を開きます。やることのプルダウンから「重なった波形を分離する」(この名称は今後変更されるかもしれません)を選択します。
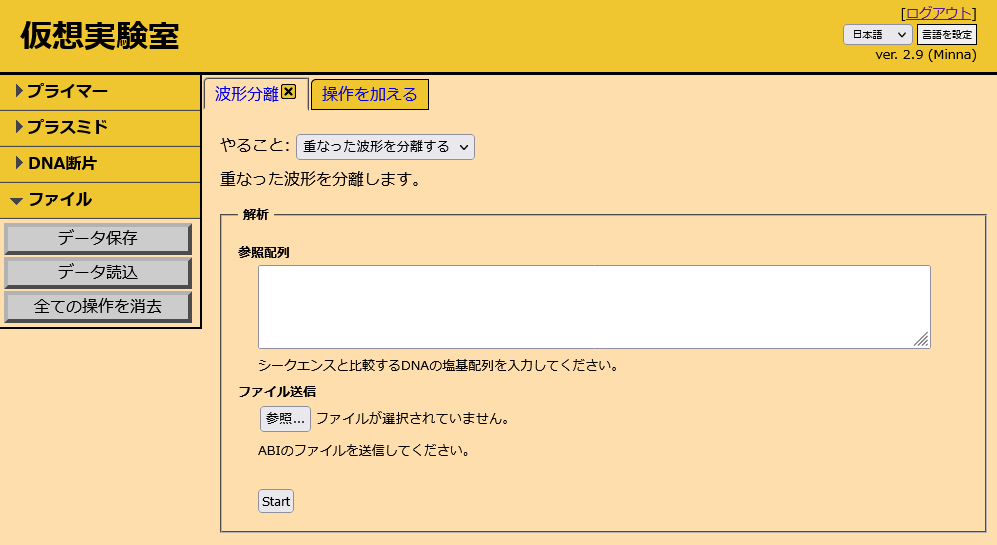
- 塩基配列を参照配列にコピーします。シークエンスデータ(ab1ファイル)を送信します。
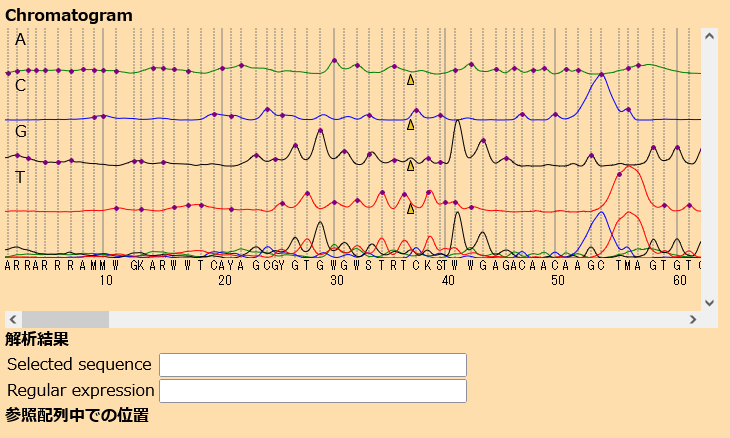
- 波形データのうちきれいに読めているところを見てみます。まず200番目ぐらいのところの塩基をクリックします。次にそこから少し離れたところをクリックすると、その間の塩基配列が選択されます。このプログラムでは塩基配列の選択方法が特殊なのでちょっと注意してください。すでに選択されたところの外側をクリックするとそこまで選択が延長されます。選択範囲をクリックするとその一塩基だけが選択されます。通常のドラッグでは選択できないので注意してください。
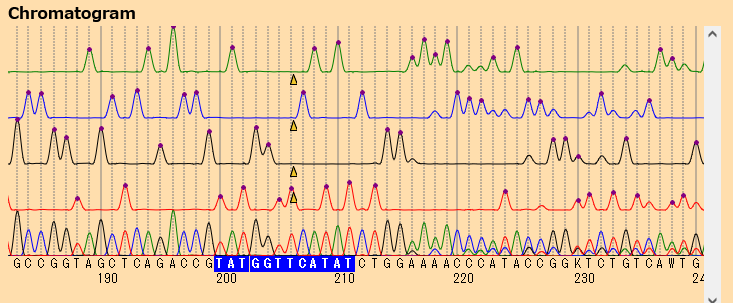
- 塩基配列を選択すると、それを参照配列中に探しにいきます。下の解析結果にあるfound1が見つかった配列で、シークエンスの200番目から214番目の塩基配列が参照配列の153番目から168番目にあることがわかります。とりえあず今はcounter1は無視してください。

- 次に塩基配列が重なって読めていないところを調べます。例えば221番目はAの波形が重なっていますが、ここに点がついていません。このAのピークをクリックします。
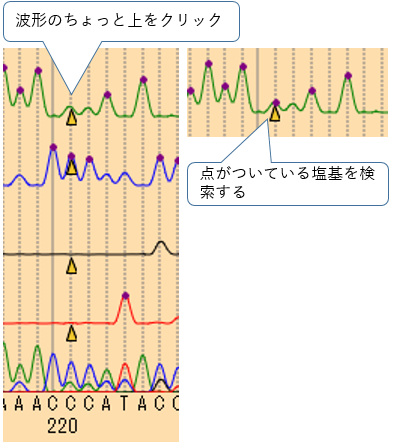
- 同様に222番目のA、223番目のC、224番目のC、と順にピークはあるけど、無視されているものをクリックしていきます。
- 229番目のGまでピークの追加を終えたら、221から229の塩基を選択します(221番目のCの文字をクリックして、次に229番目のGをクリック。途中で別の範囲が選択されたら、その中をもう一度クリック)。
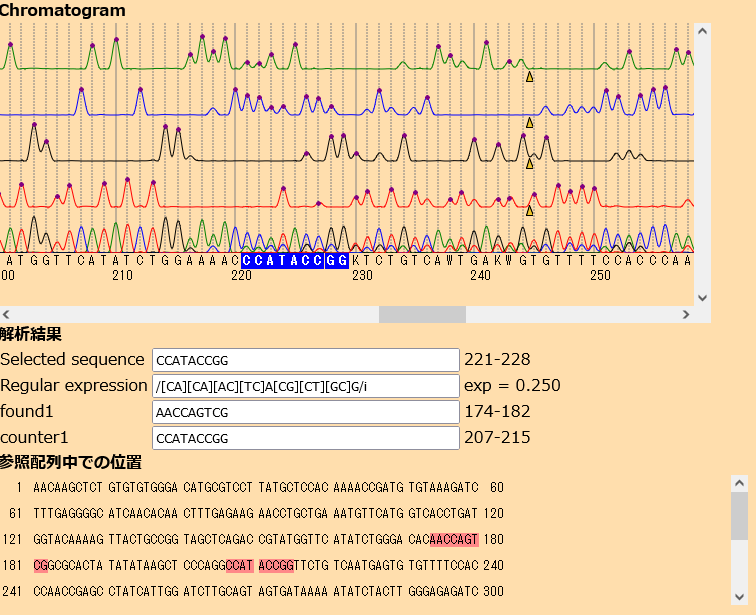
- 解析結果のfound1にAACCAGTCGが表示されます。これは参照配列の174番目から182番目と一致します。この配列を重複から引いた残りがCCATACCGGという配列で、これが207番目から215番目と一致します。シークエンスの214番目のGが参照配列の168番目と一致するので以下のようになっていると推測されます。
参照配列:TTCATATCTGGGACACAACCAGTCGGCGCACTATATATAAGCTCCCAGGCCATACCGGTTCTGTCAATGAGTGTG アリル1: TTCATATCTGGxAxACAACCAGTCG アリル2: TTCATATCTGGxAxAC------(30塩基ぐらいの欠失)-------CCATACCGG
注意
- 今のところ相補鎖の検索はできません。シークエンス方向に合わせて参照配列を用意してください。
- ギャップを考慮した検索はできません。上記の例でシークエンスの215番あたりの塩基を含んで検索をかけると参照配列を見つけることができません。CRISPR-Cas9の切断位置付近から少し離れたところで一致点を探して、そこから徐々に一致しなくなるところまで延ばしていくとよいでしょう。
- 挿入配列を見つけるのも難しいです。挿入はたぶんそれほど長くないでしょうから、50塩基ぐらい下流から検索を開始して、参照配列と一致しなくなる場所まで延ばしてください。
似た機能のプログラム
ええ、もちろんMitsucalの機能拡張の目途がついてから見つけてしまいました(車輪の再発明万歳)。
POLY PEAK PARSER
野生型と欠失のヘテロ接合のシークエンスが解析できるっぽい。
CRISP-ID
今は動いていなさそう。